Alcohol
Alcohol is one of the world’s favorite intoxicants. It is frequently found at social gatherings because it provides an anxiety-reducing effect. Consuming too much alcohol will result in alcohol poisoning, which can be fatal.



Alcohol is most often used for

Last Updated:September 28, 2022




Alcohol, otherwise known as Drinking alcohol or ethanol, is the main ingredient in a wide range of social drinks and the reason for the differentiation between 'Alcoholic' and 'Non-alcoholic' drinks.
It is a neuroactive chemical that is able to change one's perception, and has a unique metabolic pattern relative to other sources of calories (the macronutrients; carbohydrates, proteins, and dietary fats). It contributes 7 calories per gram energy-wise, but this does not always correlate well with bodyweight like the other macronutrients do.
- Ethanol
- Drinking alcohol
- Beer
- Gin
- Vodka
- Wine
- Jager
- Alcohol (The chemistry definition)
Abstinence from drinking is defined as having no ethanol intake whatsoever.
"Moderate" drinking in the literature is dependent on gender and not ultimately defined, but an upper limit can be placed at 9 units per week for women and 12-14 units a week for men, with no single event exceeding 4 units.
A unit is typically 12 oz (355 mL) of 5% beer, 5 oz (150 mL) 12.5% wine, or 0.85 oz (25 mL) of drinks with a higher (40%) alcohol content.
A recent study shows that a compound in beer may help with fat loss.
Principle Study: Matured hop extract reduces body fat in healthy overweight humans: a randomized double-blind, placebo-controlled parallel group study.
Obesity is an increasingly global problem that is associated with a greater risk of developing disorders like hypertension and diabetes. Although dieting is an effective strategy, many people find it difficult to maintain and look for easier alternatives.
A popular alternative approach to effective weight management is supplementing therapeutic products that offer ‘fat-burning’ properties. These includes natural products and ‘functional foods’ that are claimed to suppress energy intake or actively increase energy expenditure. There are many commercial products that supposedly assist in effective weight management, including compounds like conjugated linoleic acid and pyruvate, as well as natural food products such as Irvingia gabonensis and chia seed. However, most studies on these products have been inconclusive (such as for Irvingia gabonensis) or shown that these dietary supplements do not assist weight loss (such as chia seed).

On a positive note, there is promising data on the anti-obesity effects of compounds called isohumulones, or iso-α-acids. These compounds are the major bitter components in beer and come from the female hop plant (Humulus lupulus L.). As shown in Figure 1, iso-α-acids are converted from α-acids during brewing, and impart flavour and bitterness to beer. These iso-α-acids have been shown to help obese individuals with pre-diabetes by reducing hyperglycemia and body fat content. In addition, iso-α-acids have also been shown to prevent diet-induced obesity in two different strains of mice. However, one drawback of using iso-α-acids is their very strong bitter profile, which makes them quite unpalatable at the concentrations required to be effective. Although an isohumulone pill would bypass these palatability issues, for reasons unknown, it has not been widely considered.
When beer is stored for long periods of time, there is a progressive breakdown of the iso-α-acids into more complex bitter compounds—known as matured hop bitter acids (MHBA). The MHBA compounds consist of oxidised derivatives that have similar structures to iso-α-acids but are less bitter and therefore offer a more palatable therapeutic agent. Recently, it has been shown that MHBA reduces body fat in rodents at least in part by increasing thermogenesis in brown adipose tissue. Brown adipose tissue is abundant in rodents and is important for their adaptation to cold environments. Adult humans have also been shown to have metabolically active brown adipose tissue, so this may be a possible target for anti-obesity therapies in humans.
The predominant source of bitterness in beer is from the α-acid compounds present in hops. These compounds break down into iso-α-acids during brewing and in isolation may provide benefits that reduce body fat in animals and humans.
Who and what was studied?
This study investigated the potential for MHBA to reduce body fat in healthy overweight humans. Over the course of 12 weeks, participants were given a matured hop extract (MHE) that contains 18.3% MHBA with no detectable amounts of α-acids or iso-α-acids. The MHE was in the form of a test beverage that was consumed once per day and then compared against a placebo beverage of similar taste and appearance.
Researchers recruited Japanese males and females age 20-65 with a BMI of 25-30. This is classified as obesity level 1 in Japan but is classified as overweight by the World Health Organization (WHO). The participants were randomly divided into two groups: the active group and the placebo group. The active group was to consume MHE once per day for a 12-week testing period while the placebo group consumed a placebo beverage that was of similar taste and appearance.
All participants had to adhere to strict criteria that included the exclusion of any diets or dietary supplements, medication that affected fat or lipid metabolism, and excessive alcohol consumption or foods enriched in hops. They were excluded if they had any current metabolic disorders or other serious diseases, such as diabetes or heart disease. Participants were interviewed regarding their lifestyle to ascertain eligibility and compliance. They recorded daily calorie intake, physical activity and subjective symptoms. By the end of the test, evaluation of non-compliance resulted in the final numbers of 91 participants in the active group and 87 participants in the placebo group.
Hop pellets were heated to 60 degrees for 120 hours to oxidize the α-acids into iso-α-acids. Oxidized hop pellets were soaked in water at 50 degrees for one hour to extract these iso-α-acids before concentration. The liquid was then heated to 90 degrees for four hours to degrade the iso-α-acids into MHBA. The resulting matured hop extract contains 18.3% MHBA with no detectable α-acids or iso-α-acids, as judged by chromatographic analysis. Test beverages were 350 millilitres in volume and contained 35 milligrams MHBA.
Anthropometric parameters such as height, weight, waist, and hip circumferences were measured, as well as circulatory parameters such as blood pressure and pulse rate. Body fat ratio was measured through bioelectrical impedance analysis, while visceral, subcutaneous and total fat were measured through CT scanning. Blood chemistry and urinalysis were done to evaluate possible adverse effects of consuming MHE.
This study was a randomized double-blind placebo-controlled analysis of MHE consumption in healthy individuals who were classed as Japanese obese level one (or overweight according to the WHO). The hypothesis of this study was that MHE ingestion would reduce abdominal fat, BMI, waist circumferences and hip circumferences. Toxicology analysis included parameters such as blood chemistry, haematology and urinalysis to determine if MHE consumption had any adverse effects.
What were the findings?
The main study results are shown in Figure 2. Healthy obese individuals who consumed MHE had a significant reduction of five square centimetres of visceral fat and nine square centimetres of total fat area in abdominal areas after 12 weeks when compared to the placebo group. The reduction in visceral and total fat areas was approximately twofold lower in the active group than in the placebo group.
BMI and bodyweight was also significantly lower in the active group than in the placebo group, though the change from baseline was small. There was approximately a 0.5 kilogram bodyweight change in the active group compared to no bodyweight change in the placebo group after 12 weeks of MHE consumption. Waist and hip circumferences were also significantly lower in the active group when compared to baseline, with approximately one centimetre and 0.7 centimetres lost, respectively. However, the placebo group lost approximately 0.5 centimetres from the waist and hip circumferences and so there was no significant differences between the two groups.
Safety endpoint analyses showed that there was no significant variation in blood pressure throughout the entire study and, except for the four-week pulse rate measurement in the active group being higher, circulatory parameters did not change from baseline throughout the study. Blood chemistry and urinalysis showed that there were no continuously significant differences between the two groups or consistently abnormal variations from baseline. All values recorded were within normal physiological reference ranges.
The researchers also assessed subjective and adverse effects during the study to determine the safety of MHE consumption. The active group had 25 cases of cold-like symptoms during the trial, but the placebo group also reported 20 cases of cold-like symptoms, so it is unlikely that MHE causes cold-like symptoms. Other reports such as stomach ache, diarrhea, heartburn, nausea, and vomiting were also reported to study investigators. In total, there were 14 cases in the active group and 17 cases in the placebo group, suggesting that there were probably no adverse side effects to digestive parameters that could be attributed to continuously ingesting MHE.
This study investigated the effect of consuming matured hop extract for 12 weeks and whether it would reduce body fat in otherwise healthy overweight participants. After 12 weeks of MHE treatment, visceral fat area was significantly reduced when compared to the placebo group.
What does the study really tell us?
This study shows that continual consumption of MHE causes a significant reduction in body fat in healthy overweight individuals without lifestyle changes like increased physical activity or a reduction in calories consumed. The effect of the MHE on body fat reduction is likely due to the MHBA content of these beverages.
However, it is noteworthy that the placebo group also lost significant body fat—although not as much as the active group. It is possible that the placebo effect has unconsciously affected the energy balance of the participants, as has been observed in other studies on anti-obesity agents, like lactoferrin and Pueraria flower. Alternatively, seasonal variation may also affect weight, which have contributed to weight changes observed in the placebo group.
Although the test beverage contained bitter compounds, the MHBA concentration was low enough to not give the test beverage a bitter taste. However, the body has nutrient-sensing receptors in the gut, which have the ability to sense the luminal content of the stomach. This gastric sense allows the gut to initiate an appropriate response depending on the nutrients or toxins present. The gut’s bitter-sensing receptors modulate the secretion of the hunger hormone, ghrelin, when bitter compounds are ingested. It was shown that the release of ghrelin stimulates the appetite in the short-term but actually causes a long-term reduction in food intake, thus effectively reducing overall energy intake. Bitter compounds were also shown to modulate satiety by altering intestinal motility—delaying gastric emptying—thus prolonging satiety and reducing further energy intake.
However, in this study, reduced energy intake was not observed because participants were instructed to record all calories consumed, as well as maintaining their current lifestyle. Therefore, MHE-induced reduced energy intake—either by reduced appetite or increased satiety—cannot be used to explain the observed decreases in body fat in the active group. Although food intake measurement is notoriously inaccurate, the active group did consistently consume more calories than the placebo group throughout the test period.
Instead, it is suggested that MHE may accelerate energy expenditure, rather than inhibiting energy intake through the control of appetite or satiety. MHBA has been reported to enhance thermogenesis in brown adipose tissue in rodents. It does this by binding to bitter taste receptors in the GI tract. This receptor binding was shown to cause the downstream activation of sympathetic nerve activity that regulates energy and glucose homeostasis in brown adipose tissue.
Finally, this study was carried out by employees of the Kirin Company, a global beer company that also has business interests in pharmaceuticals and other health-related products.
The consumption of MHE over the course of the study resulted in a significant loss of body fat, which may have been due to the acceleration of energy expenditure rather than reduced appetite or increased satiety.
The big picture
Obesity is characterised by the excessive and pathological accumulation of body fat known as white adipose tissue: fat cells specialized in the acquiring and storing of energy. The increase in visceral fat around intra-abdominal organs is a key driver in insulin resistance and the increased risk of developing type 2 diabetes. Higher visceral fat content is therefore also associated with increased risk of cardiovascular diseases and is a key target for therapeutic approaches against obesity.
An alternative approach to combating obesity is to activate brown adipose tissue. Brown adipose tissue is thermogenic—meaning it releases energy in the form of heat—and is critical for maintaining core body temperature in small mammals, as well as in newborn human infants. New research would appear to suggest that human brown adipose tissue, when activated, has a significant impact on energy balance and body weight. In this particular study, it was shown that there is an inverse correlation between BMI and amount of brown adipose tissue in an individual. However, this observation could be explained by obese individuals being less thermally challenged due to higher mass and better insulation. This knowledge could potentially be used to develop novel therapeutic interventions that effectively reduce obesity through activation of functional brown adipose tissue.
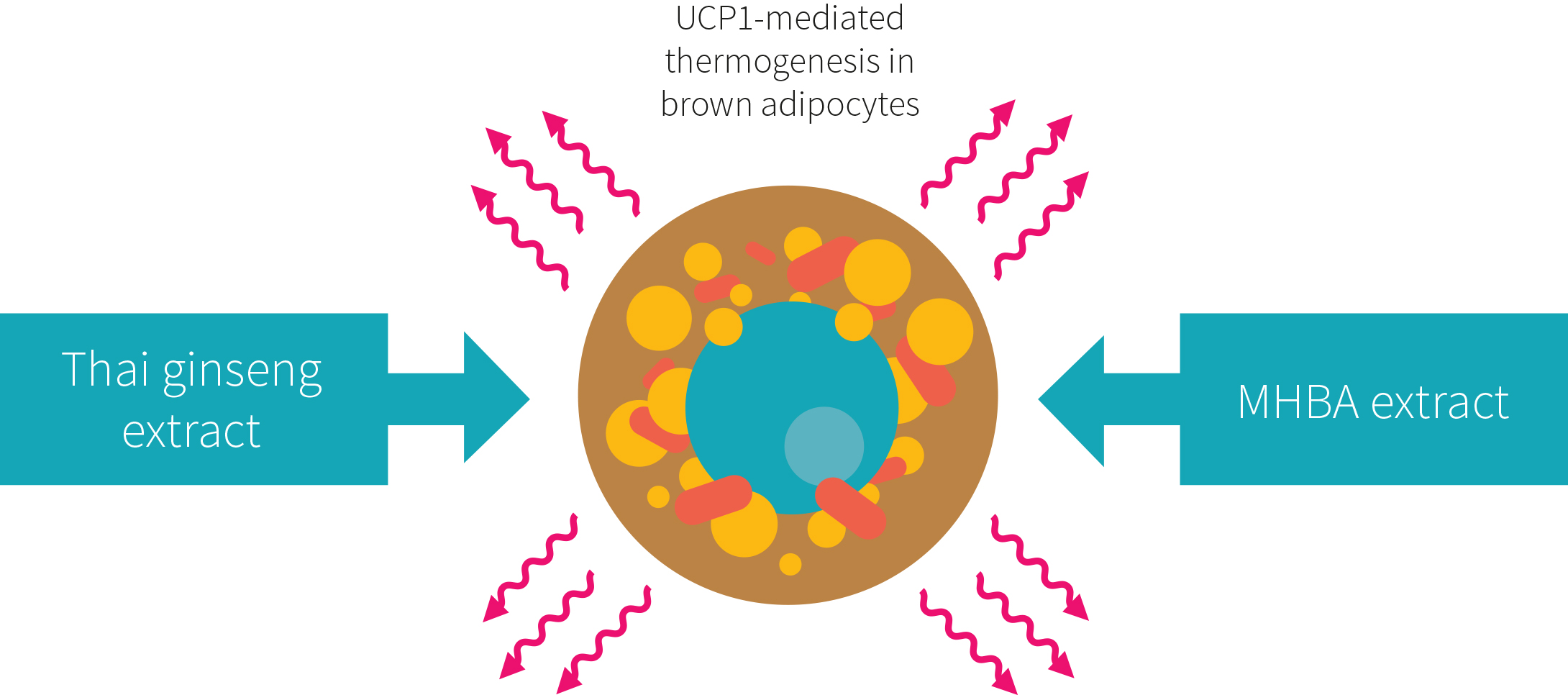
Over the years there have been several studies on the health benefits of consuming hop-derived iso-α-acids, including regulation of glucose metabolism, anti-inflammatory activity and amelioration of blood lipid profiles. More recently, matured hop bittering components were shown to influence brown adipose tissue in rodents through activation of the sympathetic nerves that innervate it. By activating brown adipose tissue, these researchers were able to demonstrate that a functional food could be used to prevent pathological body fat accumulation.

As shown in Figure 3, hops are not the only natural product that has shown promising results as a functional food to reduce body fat. Thai Ginseng (Kaempferia parviflora) is a herbaceous member of the ginger family, commonly found in Thailand. This plant is reported to have similar properties to MHE. In one study, it was reported to increase energy expenditure through activation of brown adipose tissue in mice.
The relationship between excess fat distribution and metabolic disorders is of critical importance in the fight against obesity. Hop extracts are a potential new treatment option, as they may activate brown adipose tissue and accelerate fat loss in overweight individuals.
Frequently asked questions
Q. What are the long-term effects of MHE consumption?
This study is limited by the short duration of MHE consumption. The authors postulated that continued consumption of MHE would continue to provide these benefits because no plateau in the results were observed. However, further studies over longer periods are needed to ensure continued safety and efficacy in the long-term.
Q. What are the adverse effects of consuming MHE on a regularly basis?
During this study, participants were asked to communicate any adverse effects during the study. Although a number of individuals reported digestive issues, this was the case for both active and placebo groups. Therefore, no adverse effects could be attributed to MHE consumption at the doses tested in this study.
Q. Can I just drink beer and get the same benefits?
The simple answer is, unfortunately, no. Beer—particularly strong hoppy IPAs—may have an iso-α-acid content of 30-40 ppm (30-40 milligrams per liter). However, consumption of large volumes of beer is obviously not recommended as a strategy for weight loss due to the excessive calorie content of these beverages.
What should I know?
This randomized double-blind placebo-controlled study showed that continual consumption of a bitter extract from the hop plant reduced body fat in healthy overweight individuals. Although the reductions in BMI, body weight and waist circumferences in the active group were only significant when compared to the baseline, the loss of visceral and subcutaneous fat from the abdominal area was significant when compared to the placebo group. With no obvious lifestyle changes other than the daily consumption of 35 milligrams of MHBA, this research suggests that a hop extract safely reduces abdominal fat in overweight individuals.
Before you reach for that extra pint of beer, remember, this study looked at the fat-burning potential of the compound isolated hops (found in beer).
🚧 Under Renovation 🚧
The information in this section is slated for renovation — it will soon be transformed into a more usable (and readable!) form in the coming months. As such, the text in this section may be out of date and not up to Examine’s current standards for writing style.